Peter Ferenci, Wien
Hepatische Enzephalopathie
DEFINITION
Die hepatische Enzephalopathie ist eine metabolisch induzierte, potentiell reversible Funktionsstörung des Zentralnervensystems im Rahmen von Leberkrankheiten. Im weitesten Sinne schließt sie alle neuropsychiatrischen Syndrome, die bei akuten oder chronischen Leber-erkrankungen auftreten können und kausal durch die Leberfunktionsstörung bedingt sind, ein.
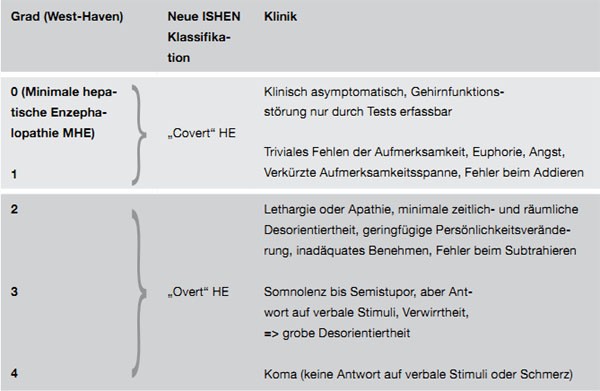
Tabelle 1: Semiquantatives Grading der Schwere der hepatischen Enzephalopathie West-Haven und ISHEN (International Society for Hepatic Encephalopathy and Nitrogen Metabolism) Kriterien
Wie schon die Vielzahl der verwendeten Synonyme und die verschiedenen Einteilungsmöglichkeiten (siehe Tabelle 1) zeigen, umfasst die hepatische Enzephalopathie (HE) ein breites Spektrum unterschiedlicher klinischer Zustandsbilder. Zahlreiche Synonyme geben meist nur Teilaspekte wieder. Voraussetzung für die Diagnose einer hepatischen Enzephalopathie ist der Nachweis einer Lebererkrankung, der Nachweis einer Funktionsstörung des zentralen Nervensystems und der sichere Ausschluss einer anderen neurologischen Erkrankung. Während der Nachweis einer Lebererkrankung bzw. die Diagnose der Funktionsstörung des zentralen Nervensystems unproblematisch ist, kann die Abgrenzung zu anderen neurologischen Erkrankungen, die bei Leberkrankheiten auftreten können, schwierig sein. Für die Praxis wichtig ist die Abgrenzung des Coma hepaticum von der Hirndrucksteigerung im akuten Leberversagen, und bei Patienten mit alkoholischer Zirrhose die Abgrenzung zu alkoholbedingten Erkrankungen des zentralen Nervensystems. Diese Abgrenzung ist für eine differenzierte Therapie notwendig. Auch für die Beurteilung der Prognose des Patienten nach einer eventuellen Lebertransplantation ist die Unterscheidung reversibler metabolischer Schäden von irreversiblen hirnorganischen Syndromen erforderlich.
PATHOGENESE
Trotz intensiver Forschung ist die Pathogenese der HE nicht erklärt. Die Gehirnfunktionsstörung könnte durch Toxine wie Ammonia, Veränderungen des Neurotransmitterstoffwechsels und der Blut-Hirn-Schranke, sowie durch proinflammatorische Zytokine hervorgerufen werden (Tab. 1). Die derzeit verwendeten „spezifischen“ Therapien basieren auf diesen verschiedenen Hypothesen der Pathogenese der HE. Unabhängig von den Hypothesen muss betont werden, daß die meisten Episoden der HE sekundäre Folge von präzipitierenden Faktoren sind. Theoretisch könnten vier Vorgänge im Rahmen des Leberversagens auftreten, die zu einer Bewußtseinsstörung führen:
a) Durch eine erhöhte Permeabilität der Blut-Hirn-Schranke könnte das Gehirn zahlreichen Neurotoxinen exponiert werden.
b) Neurotoxische Substanzen im Blut könnten die Hirnfunktionsstörungen auslösen.
c) Es könnte eine durch ein geändertes Substratangebot bedingte Störung der Neurotransmission vorliegen.
d) Der Mangel von Metaboliten (insbesondere von Glukose) oder eine Än-derung des Energiestoffwechsels die Nährstoffversorgung des Gehirns beeinträchtigen.
DIAGNOSE
Die Diagnose einer manifesten HE wird klinisch gestellt und bedarf für die Routine keiner technischen Hilfsmittel. Es gibt keinen Laborparameter, der die sichere Diagnose einer HE erlaubt. Die Erhebung der üblichen Laborparameter sollte daher dazu dienen, das Vorhandensein und die Schwere einer Lebererkrankung sowie mögliche Komplikationen des Leberversagens zu erkennen.
Spezielle Tests sind nur für wissenschaftliche Fragestellungen notwendig. Klinisch asymptomatische Patienten können aber Zeichen einer Hirnfunktionsstörung haben, die nur durch spezielle Nachweisverfahren erkannt werden können. Dieser Zustand wird als minimale (früher subklinische) HE bezeichnet. Ob es sich dabei um eine geringfügige, klinisch zu vernachlässigende Testabnormität oder um eine therapiebedürftige Krankheit handelt, ist umstritten.
Psychometrische Tests
Eine minimale HE bzw. Stadium I der HE wird durch psychometrische Tests erfasst. Die meisten Tests sind zeit- und personalintensiv und eignen sich nicht für die tägliche Praxis. Weiterhin erlauben sie keine sichere Abgrenzung zu anderen organischen Hirnschädigungen, insbesondere nicht zu alkoholbedingten Enzephalopathien. Für die Routine haben sich nur wenige Tests bewährt: Die Schriftprobe, Nachzeichentests, der Linien-Nachfahrtest und der Reitan-Test.
Die Messung der Reaktionszeit auf optische und akustische Signale sowie der Posner-Test erlauben bei kooperativen Patienten wiederholte Untersuchungen und können die zerebralen Funktions-störungen bei HE von jenen bei anderen organischen Hirnsyndromen besser diskriminieren.
EEG und neuro-physiologische Tests
Wie bei anderen Formen der Funktionseinschränkung des Gehirns eignet sich auch bei der HE die Elektroenzephalographie (EEG) zur Dokumentation und zur Quantifizierung der Schwere des Krankheitsbildes. Zur besseren Quantifizierung der EEG-Veränderungen eignen sich computergestützte Analyseverfahren wie die Errechnung dominanter EEG-Frequenzen von Intervallhistogrammen oder Frequenzprofilen.
Visuell evozierte (VEP), somatosensorisch evozierte (SEP) oder akustisch evozierte Potentiale (AEP) sowie „eventrelated“ (ER) evozierte Potentiale sind für wissenschaftliche Studien sehr hilfreich. Besonders die späte Reizantwort auf spezifische akustische Stimuli, die P300-Welle, lässt bereits eine geringfügige Hirnfunktionsstörung bei minimaler HE erkennen).
Neuere Testverfahren sind die Messung der kritischen Flimmerfrequenz und der „inhibitory control task“ Test. Diese Tests sind nicht überall verfügbar und dienen primär in wissenschaftlichen Untersuchungen zur Charakterisierung der minimalen HE.
Bildgebende Verfahren
Sofern möglich, sollte bei jedem komatösen Patienten eine Computertomographie des Gehirns durchgeführt werden. Sie erlaubt eine Abgrenzung des Krankheitsbildes von anderen neurologischen Krankheiten. Überdies kann durch die Computertomographie des Gehirns ein Hirnödem beim fulminanten Leberversagen bzw. eine Hirnatrophie bei der chronischen HE gut dokumentiert werden. Die Magnetresonanztomographie zeigt auf T1-gewichteten Bildern eine typische verstärkte Signalintensität in den Basalganglien.
THERAPIE
Das therapeutische Vorgehen wird
in Tabelle 3 zusammengefasst.
| Therapieempfehlungen bei Patienten mit Zirrhose und hepatischer Enzephalopathie |
|---|
1. Klinische manifeste HE („overt HE“) Basistherapie:
Falls keine Besserung:
Grad III-IV
|
2. Milde HE („covert HE”) Minimale HE
Stadium I
|
3. Prophylaxe bei gefährdeten Patienten (zB. nach TIPS) und zur Rezidivprophylaxe nacheiner akuten Episode
|
Tabelle 3: Therapieempfehlungen bei Patienten mit Zirrhose und hepatischer Enzephalopathie
PRÄZIPITIERENDE FAKTOREN
Tabelle 2: Präzipitierende Faktoren
- Gastrointestinale Blutung
- Sedativa oder Tranquilizer
- Verstopfung
- Dehydration / Hypovolämie durchDiuretika oder Durchfälle(Lactuloseüberdosierung)
- Hypokaliämie und / oder metabolischeAlkalose
- Hypoxie
- Hypoglykämie
Sowohl die akute wie die chronische HE ist reversibel und meist nicht die direkte Folge der Verschlechterung der Leberfunktion. In der klassischen Studie von Conn wurden bei 80 % der Fälle auslösende Faktoren wir eine gastrointestinale Blutung, eine diätetische Eiweißbelas-tung, eine hypokaliämische Alkalose, Infektionen, Verstopfung, Hypoxie oder die Gabe von Sedativa und Tranquilizern gefunden (Tabelle 2). Patienten mit fortgeschrittener Zirrhose sind besonders sensitiv für die inhibitorischen Effekte von Benzodiazepinen. Identifikation und Therapie der präzipitierenden Ursachen führt meist sehr bald zur Verbesserung der HE und ist daher die wichtigste therapeutische Maßnahme.
AMMONIAK-HYPOTHESE
Ein logisches Vorgehen zur Senkung des Blutammoniakspiegels ist die Reduktion ammoniager Substrate aus dem Gastrointestinaltrakt. Dies kann durch unterschiedliche Therapien erreicht werden: nicht metabolisierbare Disaccharide, Antibiotika und Probiotika modifizieren die Darmflora und reduzieren die intestinale Ammoniakproduktion. Weiter kann durch Verabreichung von Metaboliten des Harnstoffzyklus Ammoniak metabolisch entgiftet werden.
MODIFIKATION DER DARMFLORA
Laktulose und Laktitol
Synthetische Disaccharide können im Dünndarm nicht gespalten werden und werden von Bakterien im Kolon metabolisiert. Dabei entstehen kurzkettige Fettsäuren (Essigsäure, Milchsäure), die den pH im Kolon unter 5 senken. Im sauren Milieu entstehen Ammoniumionen, die kaum resorbiert werden. Die steigende Osmolarität wirkt katarrhtisch. Weiter wird die Kolonflora modifiziert mit Reduktion Ureaseproduzierender Bakterien. Laktulose und Laktitol werden nach wie vor als Basistherapie der HE verwendet. Etwa bei 70 bis 80 % der Patienten bessert sich die HE unter Laktulosegabe. Trotzdem ist die Evidenz für die Wirksamkeit umstritten ist. Ein Cochrane Review älterer Studien zeigte, dass Laktulose effektiver als Placebo ist, aber keinen Effekt auf die Mortalität hat. Wenn nur die methodisch einwandfreien Studien analysiert wurden, war die Änderung der HE durch Laktulose nicht unterschiedlich von Plazebo. In einer rezenten randomisiert kontrollierten Studie verbesserte Laktulose bei Patienten mit MHE die Lebensqualität und die kognitiven Funktionen. In einer anderen Studie bei Patienten, die sich von einer Episode der HE erholt hatten, senkte eine Langzeittherapie mit Laktulose die Rezidivrate.
Die Dosis von Laktulose (45 bis 90 g/d) wird individuell dem Bedarf des Patienten angepasst. Ideal sollte der Patient 2-3 weiche Stühle pro Tag haben. Überdosierung führt zu profusen Diarrhoen und kann über eine Hypokaliämie die HE verschlechtern. Laktuloseeinläufe (1-2 l 20 % Laktulose) sind in kurzer Zeit effektiv und gehört zu den Primärmaßnahmen bei Patienten mit HE.
Reduktion der Proteinzufuhr
Es gibt keinerlei Evidenz, dass eine Reduktion der Eiweißzufuhr bei Patienten mit Zirrhose sinnvoll ist. Eine Ausnahme stellen die extrem seltenen Patienten mit nachgewiesener Eiweißintoleranz dar. Ansonsten soll die tägliche Proteinzufuhr wie bei gesunden Menschen etwa 0,8/ g/kg betragen. Eine Reduktion der Proteinzufuhr führt zu einer negativen Stickstoffbilanz und erhöht die Mortalität.
Patienten mit HE Grad III/IV werden üblicherweise parenteral ernährt, nach Besserung wird die orale Eiweißtoleranz titriert. Ist eine Reduktion der Eiweißzufuhr notwendig, ist laktovegetabiles oder Fischeiweiß einer Fleischzufuhr zu bevorzugen.
Intestinale Ammoniakproduktion
Die Hemmung Urease produzierender Bakterien reduziert den anfallenden Ammoniak aus dem Darm. Seit Jahrzehnten wird Neomycin hierzu eingesetzt, obwohl bis heute kein wissenschaftlicher Beleg der Wirksamkeit vorliegt. Neuere Antibiotika wie Metronidazole, oral Vancomycin, und Rifaximin werden vermehrt verwendet. Die Wirksamkeit von Rifaximin wurde vor kurzem in einer randomisiert kontrollierten Studie belegt. Die untersuchte Dosis war 2x550 mg/Tag, auf Grund eigener Erfahrungen genügen jedoch niedrigere Dosen. Neben der Verhinderung von Rezidiven nach Episoden einer HE verbesserte Rifaximin die Lebensqualität sowie die Fahreigenschaften an einem Fahrsimulator.
Neben der Senkung des Ammoniakspiegels ist die Antibiose ein spezifischer Mechanismus um Infektionen zu verhindern. Proinflammatorische Zytokine werden heute als wichtige Mediatoren der Einschränkung der Gehirnfunktion angenommen.
Andere Modifikation der Darmflora
Probiotika, Präbiotika, Acarbose (ein Hemmer der alpha Glycosidase, welcher in der Therapie des Diabetes mellitus eingesetzt wird) und fermentierbare Fasern können die HE bessern und die Blutammoniakkonzentration senken. Die Effekte wurden in kleinen Pilotstudien beschrieben, adäquate kontrollierte Studien sind notwendig.
METABOLISCHE AMMONIAKENTGIFTUNG
Ammoniak wird im Harnstoffzyklus entgiftet. Der entstehende Harnstoff wird renal ausgeschieden. Der Harnstoffzyklus ist eine spezifische Funktion der periportalen Hepatozyten.Bei der Leberzirrhose ist die Aktivität der Carbamylphosphat-Synthetase sowie der GlutamineSynthetase (die Schlüsselenzyme der Harnstoffsynthese und der Glutaminsynthese – der alternativen Ammoniakbindung) erniedrigt, sodass die Ammoniak-entgiftung vermindert ist und es zur Hyperammonämie kommt. Solange aber diese Enzyme funktionell sind, kann die Harnstoffsynthese pharmakologisch gesteigert werden. Ornithin-aspartat ist ein Aktivator der Carbamylphosphat Synthetase und der Ornithin-Carbamyl Transferase und ist ein Substrat für die Ureagenese. Ornithin (via alpha-Ketoglutarate) und Aspartat steigern die Ammoniakentgiftung über Stimulierung der Glutaminsynthese.
Mehrere kontrollierte Studien belegen den Benefit der intravenösen Gabe von Ornithin-Aspartat gegenüber Plazebo bei Patienten mit manifester HE. Die übliche Dosierung beträgt 20 g über 4 Stunden infundiert pro Tag. Die Wirksamkeit der oralen Gabe von Ornithin-Aspartat (3x6g/d) ist weniger gut dokumentiert. Insbesondere bei der minimalen HE fehlen kontrollierte Studien. Unkontrollierte Fallserien zeigen aber eine Besserung der Lebensqualität. Ornithin-Aspartat ist beim akuten Leberversagen wirkungslos, vermutlich weil funktionell intakte Leberzellen zur Metabolisierung fehlen.
Eine andere Möglichkeit zur metabolischen Ammoniakentgiftung ist die Gabe von Natrium-Benzoat. Hierbei kommt es zu einer Bindung von Glyzin und es entsteht Hippurat, welches renal ausgeschieden wird. Diese Therapie wird in Europa nicht angewandt.
„FALSCHE NEUROTRANSMITTER“
In der Hypothese der „falschen Neurotransmisster“ wird angenommen, dass durch eine Änderung der Relation von verzweigtkettigen und aromatischen Aminosäuren im Blut vermehrt Phenylalanin ins Gehirn aufgenommen wird, wo es unter anderen zur Synthese von falschen dopaminergen Neurotransmittern wie Octopamin kommt. Das Ziel einer Therapie ist, durch die Anhebung der Konzentration der verzweigtkettigen Aminosäuren dies zu verhindern. Diese Hypothese ist umstritten. Mehrere kontrollierte Studien in den 80ger-Jahren haben diesen Therapieansatz untersucht. Die Datenlage für die intravenöse Gabe von verzweigtkettigen Aminosäuren oder von Aminosäuregemischen mit erhöhtem Anteil an verzweigtkettigen Aminosäuren ist inkonsistent. Diese Therapie wird daher heute kaum mehr verwendet.
Ebenfalls ungeklärt ist der Benefit der oralen Supplementation der Diät mit verzweigtkettigen Aminosäuren. Die einzige sinnvolle Anwendung ist bei Patienten mit nachgewiesener Eiweißunverträglichkeit gegeben. Durch Gabe von 2060 g von verzweigtkettigen Aminosäuren/Tag konnte die Eiweißzufuhr ohne Verschlechterung der HE bis auf 80g gesteigert werden
GABA-HYPOTHESE
In der GABA-Hypothese wird angenommen, dass beim Leberversagen das inhibitorische GABA-erge Neurotransmittersystem aktiviert ist. Der GABA Rezeptorkomplex besteht aus Bindungsstellen für GABA, Barbiturate und Benzodiazepine. Ziel der Therapie ist es, die inhibitorische Aktivität pharmakologisch durch Benzodiazepinantagonisten zu beeinflussen.
Der Benzodiazepinantagonist Flumazenil wurde in mehreren randomisiert kontrollierten Studien untersucht. Diese Studien zeigen, daß Flumazenil (1-5 mg iv) die HE bei mehr als der Hälfte der Patienten bessert, dass aber die Besserung nur kurze Zeit anhält. Deshalb hat sich Flumazenil nicht als Therapiestandard durchgesetzt. Hilfreich ist Flumazenil bei Patienten, die mit Benzodiazepinen behandelt wurden (z.B. Sedierung vor Gastroskopie wegen einer Varizenblutung).
Verschiedene andere Therapien wurden an kleinen Patientenzahlen untersucht, wie Zinksubstitution, Gabe von L-Carnitin, spezifische Antagonisten für Neurotransmitter (Serotonin, Glutamat, Opiate). Diese Substanzen werden aber in der Routine nicht eingesetzt.