Christoph D. Spinner1, Wajima
Safi1, Christiane Schwerdtfeger1, Dieter
Hoffmann2, Hanaa Gaber2, Fabian Geisler1, Roland M. Schmid1, Andreas
Umgelter1, München
Therapieversagen bei genotypisch nachgewiesener Resistenz
Fallbericht 1
Eine 50-jährige Patientin stellte sich zur Einleitung einer
antiviralen Tripeltherapie (DAA) bei vergeblich vorbehandelter chronischer
Hepatitis-C-Infektion (Genotyp 1b) im Herbst 2011 vor. In der Vorgeschichte
hatte eine Menghini-Punktion der Leber 2001 einen beginnenden zirrhotischen
Umbau gezeigt. Ein 2004 erfolgter Versuch einer Therapie mit
Consensusinterferon schlug nach viralem Durchbruch fehl. Ein erneuter Therapierversuch
mit Interferon und Ribavirin 2005 blieb nach initialem Abfall der HCV RNA unter
die Nachweisgrenze zu Woche 8 ebenso vergeblich. Die Viruslast stieg im Verlauf
auf max. 27 Mio. IE/ml an. Trotz fortgeschrittenen Leberschadens wurden die
Vortherapien ohne schwere Nebenwirkungen toleriert. Eine im Rahmen der dualen
Therapie aufgetretene Hyperthyreose konnte gut mit Carbimazol beherrscht
werden.
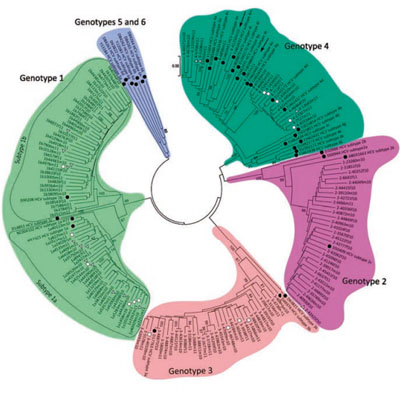 Abbildung 1: Phylogenetische Analyse von HCV bei HCV-Proteasehemer-naiven Patienten.
Abbildung 1: Phylogenetische Analyse von HCV bei HCV-Proteasehemer-naiven Patienten.
Schwarze Punkte ● verfügbare Referenzsequenzen;
graue Dreiecke ▲, Proteaseresistenz bei Baseline;
weiße Vierecke □ , HIV/HCV-Koinfizierte.
Zum Vorstellungszeitpunkt bestanden klinische und sonografische Zeichen einer kompensierte Zirrhose (Child Pugh B) mit Splenomegalie ohne Zeichen einer portalen Dekompensation. Laborchemisch waren die Transaminasen auf das 7-fache der Norm erhöht, die Lebersynthesefunktion war eingeschränkt (INR 1,2, Cholinesterase 3354 U/l), es bestand eine Hyperbilirubinämie (2,5 mg/dl) und ausgeprägte Thrombopenie (57 G/l).
Am 19.12.2011 wurde bei eine antivirale Tripletherapie mit gewichtsadaptiertem Ribavirin (15 mg/kgKG), pegyliertem Interferon alpha-2a und Telaprevir initiiert. Hierunter kam es rasch zu tolerablen Nebenwirkungen wie Juckreiz, Müdigkeit und Abgeschlagenheit. Im Verlauf der ersten Wochen traten erstmalig prätibiale Ödeme auf. Insgesamt konnte jedoch die Therapie ohne schwere Nebenwirkungen fortgeführt werden. Die Veränderungen der Laborwerte erforderte keine Dosisanpassung der Therapie. Nach einem Abfall der initialen HCV-Last (PCR) von 95.000 IU/ml (Therapieinitiierung) auf 55 IU/ml (Therapiewoche 2) und <12 IU/ml (TW 4 und 8) ergab sich zunächst der Eindruck eines Therapieansprechens. Bei in Therapiewoche 12 deutlich angestiegener HCV-Last (1.980 IU/ml) wurde die Therapie wegen primären Nichtansprechens abgebrochen. Im Verlauf konnten mittels genotypischer HCV-Resistenzbestimmung zwei relevante Mutationen (V36L und R156T) mit bekannter Kreuzresistenz für HCV Proteaseinhibitoren der 1. Generation (Telaprevir und Boceprevir) nachgewiesen werden.
Aktuell sind für diese Patientin trotz hoher Therapiedringlichkeit keine weiteren Therapieoptionen verfügbar. Bei deutlich erhöhtem alpha-Fetoprotein (AFP)-Werten (50 ng/ml) und evidenter HC-Virämie sowie dem Vorliegen einer Leberzirrhose werden derzeit in Ermangelung anderer Optionen halbjährliche Kontrollen der Leberfunktion sowie ein sonografisches HCC-Screening durchgeführt.
Fallbericht 2
Die 59-jährige Patientin stellte sich aufgrund einer chronischen Hepatitis C (Genotyp 1a, ED 1999), Transmission a.e. im Rahmen einer Bluttransfusion 1983, an unserem hepatologischen Zentrum vor. Bei langer Erkrankungsdauer lag bei der Patientin bereits eine höhergradige Leberfibrose (Fibrose Grad 3 nach Metavir) vor. In der Vorgeschichte erfolgten zwei Therapieversuche. 2000 wurde eine Therapie mit Interferon und Ribavirin begonnen, die bei Non-Response abgebrochen wurde. 2004 erfolgte ein Therapieversuch mit Consensusinterferon und Ribavirin im Rahmen einer klinischen Studie. Hierbei kam es nach initialer Virussuppression zum Relapse.
An Begleiterkrankungen bestehen eine 3-Gefäß-KHK sowie ein arterieller Hypertonus, weswegen die Patientin eine Dauermedikation mit ASS, Pravastatin und Carvedilol einnimmt.
Nach ausführlicher Beratung wurde eine antivirale
Tripletherapie mit pegyliertem Interferon, gewichtsadaptierten Ribavirin (15
mg/kgKG) und dem Proteaseinhibitor Telaprevir bei fortgeschrittener Fibrose der
Leber eingeleitet. Bei inadäquatem Abfall der Ausgangsviruslast von 1,6 Mio
IU/ml auf 210.000 IU/ml zu Therapiewoche 4 wurde die antivirale Therapie wegen
Nicht-Ansprechen unverzüglich abgebrochen. Eine Sequenzierung des Virusgenoms
ergab den Nachweis der relevanten Mutation V36M (Abb. 2), einer bekannten
Resistenzmutation für Telaprevir, mit bekannter Kreuzresistenz für Boceprevir.
Somit können alle aktuell am Markt verfügbaren Proteaseinhibotoren nicht
eingesetzt werden. Gleichzeitig wurde die Mutation R155K nachgewiesen, welche
mit einer Resistenz gegen die in Entwicklung befindlichen makrozyklischen
Proteaseinhibitoren verbunden ist. Es bestehen zum jetzigen Zeitpunkt keine
sinnvollen Therapieoptionen außerhalb klinischer Studien, weshalb neben
regelmäßigen Leberfunktionskontrollen und sonografischem HCC-Screening keine
Therapieangebote gemacht werden können.
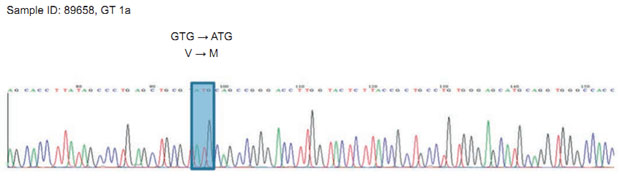 Abbildung 2: Das Chromatogramm zeigt einen Ausschnitt aus der NS3 Protease. Blau markiert ist das mutierte Codon ATG, das zum Aminosäureaustausch V → M führt.
Abbildung 2: Das Chromatogramm zeigt einen Ausschnitt aus der NS3 Protease. Blau markiert ist das mutierte Codon ATG, das zum Aminosäureaustausch V → M führt.
Fallbericht 3
Im Rahmen einer seit geraumer Zeit bestehenden Behandlungsplanung der HCV-Therapie stellte sich der 41-jährige HIV/HCV-koinfizierte MSM-Patient Anfang 2012 erneut an unserem interdisziplinären HIV-Zentrum am Klinikum rechts der Isar (IZAR) vor. Neben einer HIV-Infektion im CDC-Stadium A3 (ED 09/2007) wurde zeitgleich eine chronische HCV-Infektion GT1a, bislang therapienaiv, diagnostiziert. In der Vorgeschichte sind keine opportunistischen Infektionen bekannt, eine 12/2009 bestehende Lues-Infektion mit Lues-induzierter Vaskulitis der Augen wurde antimikrobiell therapiert und heilte im Verlauf aus. Die HIV-Infektion wurde bereits seit 2009 mit Atripla (TDF/FTC/EFV) behandelt, worunter sich eine deutliche Verbesserung der CD4-Zellen sowie eine Suppression der HIV-Last unter die Nachweisgrenze (< 40 Geq/ml) ergab. Bei geplanter HCV-Therapie war die antivirale Therapie bei Schichtdienst und hohem Wunsch des Patienten nach einer einmal täglichen Therapie auf Truvada (TDF/FTC) und Norvir-geboostetes Reyataz (ATV/r) bei guter Verträglichkeit umgestellt worden. Aktuell ergaben sich sonografisch keine Hinweise eines beginnenden zirrhotischen Umbaus der Leber oder einer Splenomegalie. Laborchemisch bestand eine Thrombopenie (102 G/l) sowie eine a.e. Atazanavir-assoziierte Hyperbilirubinämie bei moderat auf weniger als das 2-fache der oberen Normgrenze, erhöhten Transaminasen.
Nach ausführlicher Aufklärung erfolgte auf dringenden Patientenwunsch trotz fehlenden Zeichens des Leberumbaus und mangelhafter Datenlage zur Therapie der HIV/HCV-Koinfektion der Versuch einer direkten antiviralen HCV-Therapie. Zum Zeitpunkt des Beginns der HCV-Tripletherapie (DAA) mit gewichstadaptiertem Ribavirin (15 mg/kgKG), pegyliertem Interferon alpha-2a und Telaprevir ergab sich eine stabile immunologische und virologische Situation (CD4-Zellen 340 /µl, 35%, CD4/CD8-ratio 1.0, HIV-Last nicht nachweisbar). Neben Müdigkeit und Belastungsdyspnoe traten keine schwerwiegenden Nebenwirkungen auf. Die HCV-Last fiel von 7.300.000 IU/ml zum Therapiestart auf 1580 IU/ml ab und erfüllte damit nicht die Kriterien des primären Therapieansprechens der HCV-Therapie. Die Therapie wurde in Rücksprache mit dem Patienten unverzüglich abgebrochen. Die HIV-Last blieb während der gesamten Therapiedauer stabil supprimiert, der Atazanavir-Spiegel lag mit 1683 ng/ml etwa 1,5-fach oberhalb des bekannt optimalen Wirkspiegels. In einer nachträglich an unserem Institut für Virologie durchgeführten genotypischen HCV-Resistenzbestimmung ergab sich bereits zum Zeitpunkt der HCV-Therapieinitiiertung der Nachweis der relevanten Mutationen V36L, V36M und R155K. Zum Zeitpunkt des Abbruchs in Therapiewoche 4 konnte die 1. Generations-HCV Proteaseinhibitor-relevante Mutation V36M erneut nachgewiesen werden, welche am ehesten für das primäre Nichtansprechen der Therapie verantwortlich sein dürfte. Die Bedeutung des vor Therapiebeginn erfolgten Nachweises der Resistenzmutation für makrozyklische Proteaseinhibitoren R155K könnte mögliche zukünftige Therapieoptionen weiter limitieren.
Fazit
Es lässt sich zusammenfassend festhalten, dass bei virologischem HCV-Therapieversagen in allen unseren berichteten Fällen mittels genotypischer Resistenzbestimmung Proteasinhibitor-relevante Mutationen nachgewiesen wurden. Hierbei spielen die Telaprevir-relevanten Mutationen V36M und V36L, welche mit einer Boceprevir-Kreuzresistenz verbunden sind, eine Rolle. Bei gleichzeitigem Vorliegen der Mutation R155K wird auch eine Therapie mit den in Entwicklung befindlichen makrozyklischen Proteaseinhibitoren nicht möglich sein. Virusvarianten mit einer verringerten Empfindlichkeit gegenüber Proteaseinhibitoren wurden in Phase II Studien zu Telaprevir in 2% der untersuchten Patienten gefunden.1 In einer therapienaiven Kohorte mit einem hohen Anteil HIV-coinfizierter Patienten lag der Anteil von Varianten mit einer Resistenz gegen die gegenwärtig zugelassenen linearen Proteaseinhibitoren bei 9,2%.2 Da die Mutationen bereits vor antiviraler Therapie nachweisbar sind, bietet sich bei breiter Etablierung der genotypischen HCV-Resistenzbestimmung perspektivisch möglicherweise in Analogie zur HIV-Reistenbestimmung die Option unwirksame und kostenintensive Therapie zu vermeiden. Die Bedeutung der bekannten HCV-Mutation für zukünftige, insbesondere interferonfreie oder proteaseinhibitorfreie, antivirale Therapieoptionen bleibt zum jetzigen Zeitpunkt unklar.
1. Bartels DJ, Zhou Y, Zhang EZ, et al. Natural prevalence of hepatitis C virus variants with decreased sensitivity to NS3.4A protease inhibitors in treatment-naive subjects. J Infect Dis 2008;198:800–7.
2. Vicenti I, Rosi A, Saladini F, et al. Naturally occurring hepatitis C virus (HCV) NS3/4A protease inhibitor resistance-related mutations in HCV genotype 1-infected subjects in Italy. Journal of Antimicrobial Chemotherapy 2012;67:984–7.
3. Paolucci S, Fiorina L, Piralla A et al. Naturally occurring mutations to HCV protease inhibitors in treatment-naïve patients. Virol J. 2012 Oct 24;9:245.