Michael Biermer, Berlin, Und Thomas Berg, Leipzig
Ein Jahr Tripletherapie in Deutschland
Eine Bestandsaufnahme
Neue Ära
Die durch die Tripletherapie erzielte Steigerung der
antiviralen Wirksamkeit ist beeindruckend. Für Boceprevir wie für Telaprevir
konnten in den Zulassungsstudien im Vergleich zur dualen Therapie die
Heilungsraten (=dauerhaftes virologisches Ansprechen; engl.: sustained
virological response, SVR) um 30 bis 35% gesteigert werden (Abb. 1). Aufgrund
der hohen Rate eines raschen virologischen Ansprechens (RVR, definiert als
negative HCV-RNA zu Woche 4) konnte darüber hinaus bei der Mehrzahl der
Patienten die Therapiedauer auf 24 Wochen verkürzt werden1,2. Für erfolglos vorbehandelte Patienten
ergeben sich unter bestimmten Bedingungen exzellente Chancen auf das Erreichen
einer SVR, wenn eine Re-Therapie unter Einsatz eines Proteaseinhibitors erfolgt3,4.
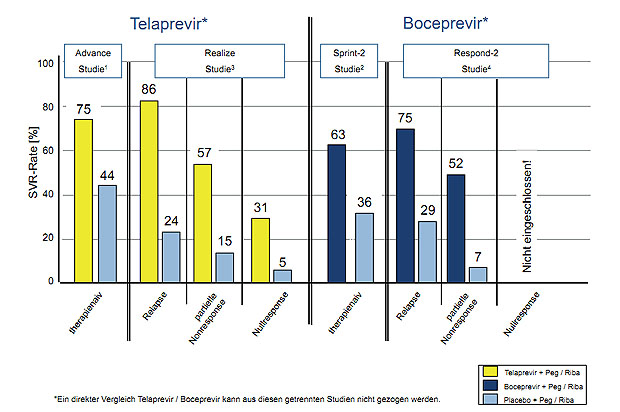 Abbildung 1: Ansprechraten in den Zulassungsstudien von Boceprevir und Telaprevir.
Abbildung 1: Ansprechraten in den Zulassungsstudien von Boceprevir und Telaprevir.
Die Zulassung von Boceprevir und Telaprevir ist beschränkt auf die Behandlung der chronischen Hepatitis C vom viralen Genotyp 1 und fordert den kombinierten Einsatz mit Peginterferon alpha und Ribavirin. Die Tripletherapie mit Proteaseinhibitor, Peginterferon alpha und Ribavirin folgt präzise formulierten Bedingungen, wobei sich die Algorithmen in Abhängigkeit vom jeweils eingesetzten Proteaseinhibitor unterscheiden. Zwingend beachtet werden müssen definierte virologische Stoppregeln, für den Fall eines ungenügenden virologischen Ansprechens.
Die vorliegende Arbeit will eine Übersicht über erste Erfahrungen nach Zulassung der Tripletherapie geben. Hierbei soll besonders der Tatsache Rechnung getragen werden, dass Therapien unter Studienbedingungen durchaus nicht immer gleichzusetzen sind mit Therapien unter „real life“ Bedingungen. Im Fokus stehen aktuell veröffentlichte „späte“ Phase 3 Studien, noch vor der Zulassung aufgelegte Programme zum bevorzugten Zugang für Patienten mit dringender Therapieindikation (engl.: early access program, EAP), multizentrisch wie monozentrisch erhobene Daten aus Patientenkohorten unter Tripletherapie sowie Studien, die relevante Aspekte für einen optimierten Einsatz der Proteaseinhibitoren liefern.
Katerstimmung?
Die ersten Monate nach Zulassung. Zweifelsohne folgte auf die Euphorie der Zulassung und der beeindruckenden Wirkungsverstärkung die Ernüchterung des erheblich gestiegenen Aufwandes in der Betreuung von Patienten unter Tripletherapie. Die Patienten müssen oft wesentlich engmaschiger betreut werden, berichten über neue, zum Teil ausgeprägte Nebenwirkungen, die Planung der Therapie umfasst jetzt die umfangreiche Abklärung potenzieller Wechselwirkungen mit anderen Medikamenten. Völlig neu ist in der Therapie der HCV-Infektion der Aspekt der virologischen Resistenzbildung.
CUPIC
In diese Zeit fiel die erste Veröffentlichung eines großen Early Access Programms (EAP) aus Frankreich, der CUPIC-Studie. Auf dem EASL-Kongress im April 2012 wurde die Interimsanalyse von den ersten 16 Wochen der Tripletherapie bei 296 Patienten unter Telaprevir und 159 Patienten unter Boceprevir vorgestellt5. Entsprechend des Charakters eines Early Access Programmes durften nur Patienten eingeschlossen werden, die einen dringenden medizinischen Bedarf für eine antivirale Behandlung hatten. Folglich waren nur Patienten mit bereits erfolgtem zirrhotischen Umbau der Leber zugelassen. Die Wahl des Proteaseinhibitors erfolgte nach Ermessen des Prüfarztes und nicht in randomisierter Form.
Die zentrale Aussage der Studie war die im Vergleich zu
allen Zulassungsstudien deutlich schlechtere Verträglichkeit der
Tripletherapie. Beinahe jeder zweite
Patient erlitt im beobachteten Zeitraum schwerwiegende Nebenwirkungen (engl.:
serious adverse events, SAE) – 144 von 296 Patienten unter Telaprevir (48,6%)
und 61 von 159 Patienten unter Boceprevir (38,4%). Bemerkenswert war, dass
insgesamt 407 SAE unter Telaprevir und 158 SAE unter Boceprevir gemeldet
wurden, so dass die meisten betroffenen Patienten zwei oder mehr schwerwiegende Ereignisse erlitten haben. Anämie trat
in beiden Kohorten sehr häufig auf, eine schwerwiegende Anämie mit Hb < 8,0
g/dl wurde unter beiden Therapieregimen in 10,1% der Fälle beobachtet. In dieser schwierig zu behandelnden Patientengruppe war die Inzidenz von Infektionen auffällig, die überwiegend zur
unerwartet hohen Mortalitätsrate dieser Kohorte beigetragen hat: Unter Boceprevir
sind in den ersten 16 Wochen der Therapie zwei Patienten verstorbenen, unter
Telaprevir sechs Patienten. Beide Todesfälle unter Boceprevir und drei von
sechs Todesfällen unter Telaprevir sind im Zusammenhang mit Infektionen und/oder septischer Reaktion zu sehen.
Zu gefährlich bei fortgeschrittener Lebererkrankung? Für die längste Zeit des Jahres 2012 standen außer den CUPIC-Daten keine weiteren Studien zur Verträglichkeit der Tripletherapie zur Verfügung. Daher wurde mit großer Spannung die Veröffentlichung weiterer Kohorten erwartet. Mit Interimsanalysen eines großen internationalen Early Access Programmes6 und zweier nicht-interventioneller Beobachtungsstudien aus Deutschland (TEPS-Studie7 und PAN-Studie8) liegen seit November 2012 weitere Daten zur Effektivität und Verträglichkeit zur Tripletherapie im klinischen Alltag vor.
Das internationale Telaprevir Early Access Programm mit über
1900 eingeschlossenen Patienten fokussiert wie die CUPIC-Studie auf Patienten
mit bereits fortgeschrittener Leberfibrose oder Zirrhose. Die jetzt
vorgestellte Analyse berichtet von den ersten 609 Patienten, die Woche 16 der
Behandlung erreicht haben. 20% der Patienten waren unvorbehandelt, bei 55% war
eine Zirrhose (F4), bei 45% eine schwere Leberfibrose (F3) nachgewiesen.
Insgesamt fiel die
Sicherheitsanalyse dieser Kohorte mit einer SAE-Rate von 14% und einer
Abbruchrate aufgrund von Nebenwirkungen von ebenfalls 14% deutlich günstiger
aus als in der CUPIC Studie, in die ausschließlich zirrhotische Patienten
eingeschlossen wurden. Erneut standen Anämie und Hautausschlag im Fokus der
Verträglichkeits-Analyse. Insgesamt traten im berichteten Zeitraum der ersten
16 Wochen Hautreaktionen bei 42% der Patienten auf, die in der Mehrzahl jedoch
leicht oder mittelgradig ausgeprägt waren und nicht zum Abbruch der Therapie
führten. Bei 4% erreichten die Hautreaktionen einen Schweregrad 3 und bei einem
Patient entwickelte ein Stevens-Johnson-Syndrom, das sich nach Absetzen der
Therapie zurückbildete. Eine Anämie (definiert als Hb < 10 g/dl)
entwickelten 59% der Patienten, davon 3% Grad 3 oder 4 (< 9 g/dL oder Abfall
von > 4,5 g/dL). Die Therapie der Anämie bestand zu 28% in einer
Ribavirindosisreduktion, 24% Einsatz von Erythropoeitin und 11.5%
Transfusionen. Auch die Mortalitätsrate war im Vergleich zu CUPIC-Kohorte
geringer (3/609 = 0.5%). Bei 2 Patienten kam es zur Dekompensation der
Leberzirrhose mit Leberversagen und bei einem Patienten trat eine fatale
ischämische Kolitis auf.
Zirrhose und dekompensierte Zirrhose
Auch eine kleinere Kohorte von Patienten mit kompensierter Leberzirrhose aus Deutschland wurde auf der AASLD-Tagung 2012 vorgestellt9. In der großen nicht-interventionellen PAN-Studie des Bundes der niedergelassenen Gastroenterologen (bng) (Zitierung Forestier et al.) wurden von insgesamt 598 Patienten mit Telaprevir basierter Tripletherapie 43 Patienten mit kompensierter Leberzirrhose identifiziert. Im beobachteten Zeitraum der ersten 12 Therapiewochen erlitten vier dieser Patienten eine oder mehrere SAE (insgesamt 7 SAE Meldungen). Bis zur Woche 12 brachen 4 von 43 Patienten die Therapie aufgrund einer Unverträglichkeit (2 Patienten) oder auf eigenen Wunsch (2 Patienten) ab. Vier Patienten erfüllten die virologischen Stoppregeln zu Woche 4 und/oder 12. Die SAE Rate von 9% ist vergleichbar mit den Ergebnissen aus den Zulassungsstudien und bestätigt die hohe Rate aus der CUPIC-Studie ebenfalls nicht.
Die Betrachtung der Verträglichkeit einer Tripletherapie vor dem Hintergrund einer fortgeschrittenen Lebererkrankung wird mit der von Verna et al. auf der AASLD 2012 vorgestellten Kohorte10 gewissermaßen auf die Spitze getrieben. 28 Patienten auf der Warteliste zur Lebertransplantation erhielten eine Tripletherapie (26 auf der Basis von Telaprevir, 2 auf der Basis von Boceprevir). Der MELD-Wert lag im Median bei 8 (Spannweite 6-16), ein Viertel der Patienten war mit Aszites dekompensiert und bei 39% lag ein hepatozelluläres Karzinom in Zirrhose vor. Die Therapie wurde bei 43% der Patienten vorzeitig beendet, ein Patient verstarb aufgrund einer Dekompensation der Leberzirrhose. Selbst in dieser Kohorte schwerkranker Patienten war die SAE-Rate mit 21% deutlich geringer als in der CUPIC-Kohorte. Ein mögliche Erklärung für die geringere Komplikationsrate ist die bei 32% der Patienten erfolgte prophylaktische Gabe von Antibiotika.
Risiko überschätzt?
Warum ist die Verträglichkeit in CUPIC so viel schlechter
als in allen anderen Studien? Abbildung 2 stellt die unterschiedlichen SAE-Raten der
vorgestellten Studien gegenüber und belegt, dass die CUPIC-Erfahrungen bisher
in keiner Studie bestätigt werden konnten. Warum aber sind die Patienten in
Frankreich so viel häufiger unter Therapie in Schwierigkeiten geraten als in
allen anderen Studien? Eine mögliche Erklärung ist, dass das Patientenkollektiv
der CUPIC-Studie doch insgesamt ein fortgeschritteneres Erkrankungsstadium
aufwies mit einem hohen Anteil bereits dekompensierter Leberzirrhose und
relevanter Begleiterkrankungen. Außerdem waren die Patienten in CUPIC die
ersten, die außerhalb der Zulassungsstudien eine Tripletherapie erhielten,
sodass in allen folgenden Erhebungen bereits eine Lernkurve im „real life“
Einsatz der neuen Substanzen abzulesen ist. Viele Krankenhausaufenthalte in der
CUPIC-Studie erfolgten zudem zur Transfusion bei ausgeprägter und rasch
aufgetretener Anämie unter Therapie. Der mittlerweile erbrachte Beweis, dass
zur Therapie der Anämie unter Tripletherapie eine Reduktion des Ribavirin ohne
Beeinträchtigung der SVR-Raten erfolgen kann (die Daten hierzu werden weiter
unten im Artikel diskutiert), trägt vermutlich ebenfalls zur reduzierten
Hospitalisierung aufgrund von Anämie in den späteren Studien bei.
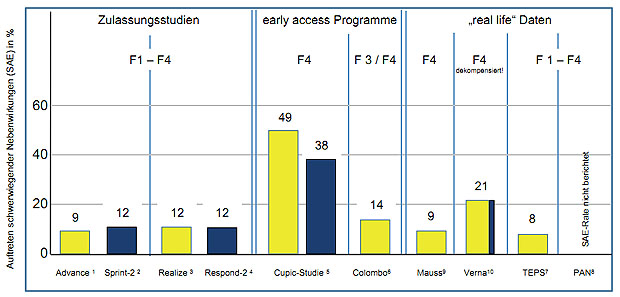 Abbildung 2: Schwerwiegende Nebenwirkungen (SAE) in Zulassungsstudien, EAP-Programmen und Kohorten aus dem Klinikalltag. Telaprevir (gelb) und Boceprevir (blau)
Abbildung 2: Schwerwiegende Nebenwirkungen (SAE) in Zulassungsstudien, EAP-Programmen und Kohorten aus dem Klinikalltag. Telaprevir (gelb) und Boceprevir (blau)
Die Theorie der noch ansteigenden Lernkurve wird letztlich auch direkt in der CUPIC-Kohorte bestätigt. Mit zunehmender Patientenzahl in der Folge-Veröffentlichung auf der AASLD Tagung 2012 ist ein Absinken der SAE Rate für beide Therapieregime zu beobachten11. Von insgesamt 497 in die Analyse eingeschlossenen Patienten mit Leberzirrhose und erfolgloser Vorbehandlung wurden 205 mit Boceprevir- und 292 mit Telaprevir-basierter Tripletherapie behandelt. Die SAE Rate während der ersten 16 Wochen der Therapie lag mit 32.7% und 45,2% geringfügig unter der ersten Analyse vom EASL 2012. Vor dem Hintergrund der zwischen beiden Analysen steigenden Patientenzahl scheint die Verträglichkeit mit zunehmender Dauer des CUPIC-Programms besser zu werden.
Tripletherapie ist wirksam!
Alle nach Zulassung erhobenen und hier diskutierten Daten beruhen auf Interims-Analysen unter noch laufender Therapie. Berichtet werden Wirksamkeit und Verträglichkeit der Therapie für einen Beobachtungszeitraum der ersten maximal 16 Behandlungswochen. Wenngleich somit noch keine SVR-Raten berichtet werden können, sollten die sehr soliden Daten zur frühen antiviralen Wirksamkeit unter laufender Therapie nicht unbeachtet bleiben:
CUPIC-Studie: Betrachtet man die gesamte Telaprevir-Kohorte (ITT-Analyse (engl.: intention to treat)), so erreichten 79% eine komplette virologische Response (HCV-RNA negativ) zu Woche 12 der Tripletherapie. In der Analyse nach Protokoll behandelter Patienten (per Protokoll-Analyse - PP) betrug die virologische Responserate zu Woche 12 93%. In der Boceprevir-Kohorte wurde eine negative HCV-RNA zur Woche 16 (also nach 12 Wochen Tripletherapie) in 58% (ITT) bzw. 77% (PP) der Fälle erreicht. Bei der Interpretation der Ergebnisse muss jedoch berücksichtigt werden, dass in der Mehrzahl Patienten mit Relapse und partieller Response in der Vortherapie eingeschlossen wurden und die Zahl der Patienten mit Nullresponse ausgesprochen gering war.11
Telaprevir-EAP: Die Analyse umfasst die ersten 12 Wochen der Therapie und beschreibt ein komplettes virologisches Ansprechen (HCV-RNA nicht detektierbar) zu Woche 12 in 85% bei therapienaiven Patienten und ebenfalls 85% bei Patienten nach Relapse in Vortherapie. Patienten mit Nonresponse (partielle oder Nullresponse) oder viralem Durchbruch unter Vortherapie erreichten eine negative HCV-RNA zu Woche 12 in 73% (alle Daten nach ITT-Analyse)6.
Zirrhose-Kohorte der PAN-Studie: Von 43 Patienten mit kompensierter Zirrhose erreichten unter Telaprevir basierter Tripletherapie 18 (41%) zu Woche 4 und 27 (63%) zu Woche 12 eine negative HCV-RNA (<10 IU/ml)9.
Dekompensierte Zirrhose-Kohorte: Zu Woche 12 der Therapie lag die Rate negativer HCV-RNA bei 71%, negativ oder nicht quantifizierbar war die HCV-RNA bei 86% der Patienten. Eine Lebertransplantation erhielten acht der 28 Patienten. Zum Zeitpunkt der Transplantation waren 7 der 8 Patienten HCV-RNA negativ. Nur bei einem Patienten kam es in der Folge nach Tx zur Reinfektion.10
Alle Fibrosestadien
Tripletherapie unter echten „real life“ Bedingungen. Alle bis hier besprochen Studien fokussierten auf Patienten mit fortgeschrittener Fibrose oder Leberzirrhose. Für ein Abbild echter „real life“ Bedingungen sollten jedoch Patienten in allen Stadien der Erkrankung betrachtet werden, weshalb die Ergebnisse zweier großer nicht-interventioneller Studien aus Deutschland, für die kürzlich Interimsdaten veröffentlicht wurden, interessant sind.
Berg et al. veröffentlichten kürzlich eine 12 Wochen-Interimsanalyse der ersten 100 Patienten (12,5% der geplanten Gesamtzahl) aus der nicht-interventionellen TEPS-Studie zum Einsatz der Telaprevir-Tripletherapie7. In dieser Kohorte wiesen lediglich 11% der eingeschlossenen Patienten eine Zirrhose auf. Zwei Drittel der Patienten waren bereits vorbehandelt. Wiederum beeindruckend waren die intialen virologischen Ansprechraten unter Therapie mit einem kompletten virologischen Ansprechen (HCV-RNA negativ) zu Woche 4 von 67% und zu Woche 12 von 91%. Neun der 100 Patienten (9%) erfüllten die virologischen Stopp-Kriterien zu Woche 4 der Therapie (HCV-RNA > 1000 IU/mL) und bei allen neun Patienten wurde die Therapie auch folgerichtig beendet. Während nahezu jeder Patient über Nebenwirkungen berichtete (psychische NW mit 57% am häufigsten, gefolgt von trockener Haut, Müdigkeit (je 54%), gastrointestinalen (48%) und anorektalen (30%) Symptomen, Hautausschlag (28%), Anämie (23%) und Infektionen (20%)), ist die SAE-Rate mit 8% im Vergleich zu CUPIC-Kohorte ausgesprochen gering.
Real Life – Boceprevir und Telaprevir
Forestier et al. stellten auf der AASLD Tagung 2012 eine erste Interim-Analyse der PAN Studie vor, einer weiteren großen nicht-interventionelle Studie aus Deutschland, die vom bng mit Unterstützung der Roche Pharma AG durchgeführt wird8. Von insgesamt 769 Patienten erhielten 195 eine Tripletherapie mit Boceprevir und 574 eine Tripletherapie mit Telaprevir. Die Baseline-Charakteristika in beiden Therapiegruppen waren bezüglich Alter, BMI, Viruslast, Anteil HCV-Subtyp 1a versus 1b, IL28B-Genotyp und Dauer der Infektion vergleichbar. Leider wurde der Anteil der Patienten mit fortgeschrittener Fibrose oder Zirrhose nicht berichtet. Wie in der TEPS-Studie waren zwei Drittel der Patienten bereits antiviral vorbehandelt. Die Auswahl des Proteaseinhibitors erfolgte einer nicht-interventionellen Studie entsprechend nicht nach Randomisierung, sondern nach dem Ermessen des Therapeuten (und/oder Wunsch des Patienten). Zum Zeitpunkt der Analyse haben 627 Patienten Woche 4, 444 Patienten Woche 8 und 330 Patienten Woche 12 der Therapie erreicht. Nicht für alle Patienten lagen HCV-RNA Ergebnisse vor, so dass die Wirksamkeitsanalyse sich nur auf Patienten mit auswertbarem Befund beschränkt. Unter Telaprevir -Tripletherapie lagen die virologischen Responseraten (negative HCV-RNA) zu Woche 4, 8 und 12 bei 62%, 78% und 74%, unter Boceprevir-Tripletherapie (bis Woche 4 nur Lead-in mit Peginterferon-alpha und Ribavirin) waren die Ansprechraten zu Woche 4, 8 und 12 bei 7%, 63% und 72%. Während unter der Lead-in Phase der Boceprevir-Therapie eine Vergleichbarkeit der Ansprechraten nicht gegeben ist, nähern sie sich bereits zu Woche 8 an, zu Woche 12 ist dann ein Unterschied der Wirksamkeit nicht mehr zu sehen.
Bis Woche 12 der Therapie haben 18 Patienten aufgrund von Unverträglichkeit und 8 Patienten auf eigenen Wunsch die Telaprevir-basierte Therapie abgebrochen (26/216 ausgewerteten Patienten – Abbruchrate 12%), in der Boceprevir Kohorte haben drei Patienten wegen Unverträglichkeit und ein Patient auf eigenen Wunsch die Therapie abgebrochen (4/114 ausgewerteten Patienten – Abbruchrate 4%). Eine detaillierte Analyse der Verträglichkeit, insbesondere des Auftretens von SAE ist in der Interimsanalyse leider nicht erfolgt, was den Vergleich zu den anderen Studien erschwert.
Boceprevir bei Nullresponse
Die PROVIDE-Studie bestätigt nachträglich die Zulassung von Boceprevir für Nullresponder. Neben der Leberzirrhose stellt vor allem die Nullresponse auf eine Interferon-basierte duale Therapie einen ungünstigen prognostischen Faktor für das Erreichen einer SVR unter Tripletherapie dar3 . So erreichten nur 31% der Nullresponder unter einer Telaprevir-Tripletherapie (REALIZE-Studie) eine SVR. Liegt bereits eine Zirrhose vor, so ist die Chance auf eine SVR nur noch 14%. In der Boceprevir Zulassungsstudie zur Re-Therapie (Respond-2) wurden Patienten mit Nullresponse in der Vortherapie nicht eingeschlossen.4 Mit der PROVIDE-Studie12, die auf der EASL Tagung 2012 präsentiert wurde, konnte nun gezeigt werden, dass auch Patienten mit Nullresponse vergleichbar effektiv mit einer Boceprevir-Tripletherapie behandelt werden können.
52 Patienten, die in den Kontrollarmen der Boceprevir
Phase-2- und 3-Studien unter Peginterferon-alpha und Ribavirin die Kriterien
der Nullresponse erfüllten (Woche 12 Viruslast Abfall < 2log10), erhielten
in der PROVIDE-
Studie (unverblindet) eine Boceprevir- Tripletherapie. 19 von 50 ausgewerteten
Patienten erreichten eine SVR, was einer Rate von 38% entspricht. Ein direkter
Vergleich der Ergbnisse der PROVIDE-Studie mit denen REALIZE-Studie (SVR 31%
unter Telaprevir-Tripletherapie) ist jedoch problematisch, da die
PROVIDE-Studie nicht unter kontrolliert randomisierten Bedingungen durchgeführt
wurde und auch der Anteil von Patienten mit fortgeschrittener Fibrose oder
Zirrhose mit 10% deutlich niedriger war als in der REALIZE-Studie (33%).
Schwieriges Dosierungsschema
Die Optimize Studie verspricht eine Erleichterung in der Einnahme von Telaprevir. Neben der Bewältigung des erweiterten Nebenwirkungsspektrums stellt die Umsetzung der Medikationseinnahme für Patienten und Therapeuten eine nicht zu unterschätzende Herausforderung dar. Beide Proteaseinhibitoren müssen genau alle acht Stunden eingenommen werden. Für Telaprevir kommt noch erschwerend hinzu, dass die Einnahme mit einer fettreichen Mahlzeit erfolgen soll. Dabei lässt sich in der Regel das Achtstunden-Intervall nicht an den normalen Mahlzeiten-Rhythmus des Patienten anpassen. Gleichzeitig gilt die strikte Einhaltung der Einnahmezeitpunkte als Grundvoraussetzung für eine erfolgreiche Therapie.
Auf der AASLD Tagung 2012 wurde mit der OPTIMZE-Studie (Phase-3b)13 der Beweis erbracht, dass eine Streckung des Einnahmeintervalls von Telaprevir auf 12 Stunden ohne Einbuße an Wirksamkeit erfolgen kann, wenn die Gesamt-Tagesdosis gleich bleibt. 740 therapienaive Patienten wurden randomisiert und erhielten eine Tripletherapie bei der Telaprevir entweder mit 750 mg alle acht Stunden oder mit 1125 mg alle zwölf Stunden eingenommen wurde. Die gesamte Telaprevir-Tagesdosis betrug also in beiden Behandlungsarmen gleichermaßen 2250 mg. Das Studienziel, die Nichtunterlegenheit der zweimal täglichen Einnahme wurde eindrucksvoll erreicht: Die SVR Rate war in beiden Armen fast identisch mit 73% (279 von 371 Patienten) im 8-Stunden-Arm und 74% (274 von 369 Patienten) im 12-Stunden-Arm. Auch bei Betrachtung besonderer Untergruppen ergaben sich keine signifikanten Unterschiede. So betrug für Patienten mit kompensierter Zirrhose die SVR-Rate 49% (alle 8 h) im Vergleich zu 54% (alle 12 h). Auch die Rate des raschen virologischen Ansprechens (RVR), und damit die Vorrausetzung für eine Therapieverkürzung auf 24 Wochen, war in beiden Armen vergleichbar (67% (alle 8 h) versus 69% (alle 12 h)).
Dass 2 x 3 also ebenso effektiv ist wie 3 x 2 (Tabletten)
wird bereits jetzt viele Therapeuten dazu bewegen, die Dosisintervalle
entsprechend umzustellen (Abb. 3). Offiziell wird es voraussichtlich ab
Frühjahr 2013 zu einer Anpassung der Zulassung von Telaprevir auf 2 x 1125 mg
kommen. Dies stellt eine wesentliche
Erleichterung im therapeutischen Management dar.
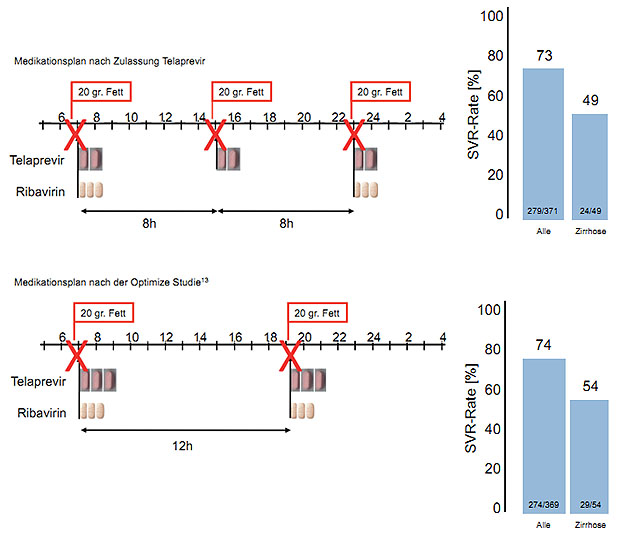 Abbildung 3: Medikationsplan für Telaprevir aktuell und in der Zukunft?
Abbildung 3: Medikationsplan für Telaprevir aktuell und in der Zukunft?
Nebenwirkungsmanagement
Wie sollen wir auf die Anämie unter Tripletherapie
reagieren? Auch unter dualer Therapie gehört das Auftreten einer Anämie zu den
häufigsten Nebenwirkungen. Die Anämie unter dualer Therapie ist jedoch meist
nicht sehr ausgeprägt (selten < 9 g/dl) und korreliert positiv mit dem
Erreichen einer SVR. Aufgrund einer klaren Korrelation zwischen Ribavirindosis
und Therapieerfolg wurde unter dualer Therapie stets versucht, möglichst hohe
Ribavirindosierungen beizubehalten.
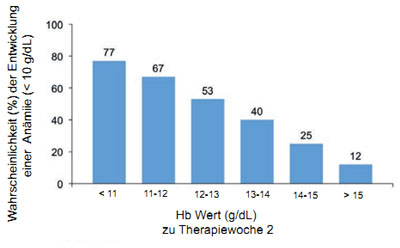 Abbildung 4: Der absolute HB-Wert zur Therapiewoche 2 als Prädikator für das Erreichen einer Anämie (<10 g/dL) im weiteren Verlauf
Abbildung 4: Der absolute HB-Wert zur Therapiewoche 2 als Prädikator für das Erreichen einer Anämie (<10 g/dL) im weiteren Verlauf
Sowohl Telaprevir als auch Boceprevir wirken zusätzlich Anämie-verstärkend. Bester Prädiktor für die Entwicklung einer Anämie unter Telaprevir-Therapie ist der absolute Hb-Wert zur Woche 2. Bei einem Hb-Wert von < 11 g/dl ist, einer post hoc-Analyse der REALZE Studie14 zufolge in 77% der Fälle damit zu rechnen, im weitern Verlauf eine signifikante Anämie (Hb <10 g/dl) zu entwickeln, während ein Hb von > 15 g/dl zu Therapiewoche 2 nur mit einer Anämie-Wahrscheinlichkeit von 11% verbunden ist (Abb. 4).
Um die optimale therapeutische Reaktion auf eine Anämie unter Tripletherapie zu ermitteln, wurden in eine große Phase-3b-Studie 678 Patienten eingeschlossen und mit Boceprevir, Peginterferon-alpha und Ribavirin behandelt. Patienten, die unter Therapie einen Hb-Wert von <10 g/dL erreichten, wurden randomisiert und erhielten entsprechend dem zugewiesenen Studienarm entweder eine Reduktion der täglichen Ribavirindosis in Schritten von 200 bis 400 mg oder 40.000 IE Erythropoetin als wöchentliche subkutane Injektion.
Insgesamt wurden 500 Patienten randomisiert (249 zu Ribavirin Reduktion und 251 zu EPO) und in beiden Armen wurde eine identische SVR-Rate von 71% erzielt15. In weitern Analysen konnte auch gezeigt werden, dass die Ribavirin-Reduktion unabhängig von ihrem Zeitpunkt, von ihrer Dauer und auch von der minimal eingesetzten Dosis ohne Einbuße der Wirksamkeit erfolgen kann 16. Wenn zum Zeitpunkt der Reaktion auf die Anämie die HCV-RNA negativ war, ist eine SVR in 86% der Fälle erreicht worden, wurde bei noch nachweisbarer HCV-RNA auf die Anämie reagiert, wurde eine SVR noch in 56% der Fälle erreicht. Erneut waren die SVR-Raten in beiden Studienarmen, also zwischen Ribavirin Reduktion oder EPO Gabe identisch (86 und 56%). Entsprechend zur Ribavirinreduktion unter Boceprevir Triple Therapie hatte eine retrospektive Analyse der Phase 3 Studien Advance, Illuminate und Realize auch für die Telaprevir basierte Triple Therapie keine signifikante Reduktion der SVR-Rate gezeigt, wenn in Reaktion auf eine Anämie die Ribavirindosis vermindert wurde17 – in den therapienaiven Kohorten (Advance und Illuminate) wurde ohne Ribavirinreduktion eine SVR in 79% der Fälle erreicht, bei Reduktion auf 800-1000 mg/d in 75% und bei Reduktion auf ≤ 600 mg/d in 74% der Fälle. Diese Ergebnisse helfen ein Dilemma zu lösen, dem Therapeuten der Hepatitis C immer wieder gegenüber standen: Erythropoetin ist in Deutschland zur Behandlung der Hepatitis C (oder Therapie-) assoziierten Anämie nicht zugelassen und die Reduktion des Ribavirin vermindert die Heilungs-Chancen. Letzteres ist für die Boceprevir wie für die Telaprevir basierte Tripletherapie jetzt widerlegt und stellt nicht nur aus Kostengründen die Strategie der Wahl zum Anämie-Management dar.
Langzeitstudien nach Tripletherapie
Stabilität einer SVR und Problematik der Resistenzmutationen. Trotz der deutlichen Verstärkung der antiviralen Wirksamkeit durch den Einsatz von Proteaseinhibitoren werden auch unter Tripletherapie Patienten mit nicht erfolgreicher Therapie beobachtet. Ganz im Gegensatz zum virologische Nichtansprechen auf die duale Therapie wird unter Einsatz der Proteaseinhibitoren erstmals in der Therapie der chronischen HCV-Infektion mit mutationsbedingter Resistenzbildung des Virus gerechnet werden. Einzelne Mutationen im Bereich des für die Protease-kodierenden Virusgenoms können zur Einschränkung der antiviralen Wirkung von Proteaseinhibitoren bis hin zum kompletten Wirkverlust führen.
Die relevanten mit Resistenz-assoziierten Virusmutationen (RAV) werden gleichermaßen für Boceprevir wie für Telaprevir in den Positionen V36A/M; T54S/A; V55A; R155K/T/Q; A156S; A156T/V; V170A/T beschrieben18. Resistenz-assoziierte Varianten sind jedoch oft im Vergleich zum Wildtyp-Virus in ihrer replikativen Fitness eingeschränkt, weshalb sich diese Varianten im Laufe der Zeit nach Ende einer Proteasehemmer-Exposition gegenüber Wildtyp-Virus nur schwer behaupten können. Dies wurde in zwei wichtigen Studien zur Langzeit-Nachbeobachtung nach Ende einer Proteaseinhibitor basierten Therapie belegt: Bereits im Jahr 2010 wurde mit der Extend-Studie19 eine Langzeit-Nachbeobachtung von Patienten nach Telaprevir basierter Tripletherapie vorgestellt. Die Studie konnte auf der einen Seite die Dauerhaftigkeit einer erreichten SVR belegen, indem 122 von 123 Patienten nach erreichter SVR in weiterer Nachbeobachtung von median 22 Monaten HCV-RNA negativ blieben. Andererseits konnte gezeigt werden, dass bei 50 von 56 Patienten (89%), die keine SVR erreichten und zum Ende der Telaprevir-Tripletherapie RAV aufwiesen, diese Mutationen im medianen Beobachtungszeitraum von 25 Monaten gegenüber Wildtyp-Virus nicht mehr nachweisbar waren 19.
Auf der diesjährigen AASLD Tagung konnten nun entsprechende
Daten auch zur Boceprevir-Therapie präsentiert werden. Barnard et al. stellten
die Studie zur Langzeit-Nachbeobachtung nach einer Boceprevir-Tripletherapie
vor20. Auch hier konnte die Dauerhaftigkeit einer erreichten SVR
belegt werden mit 691 von 696 Patienten, die im median 31 Monate nach Erreichen
der SVR weiterhin HCV-RNA negativ waren. Bei drei Patienten wurde ein
bestätigter später Relapse gesehen. Einer dieser Patienten zeigte jedoch einen
Wechsel des HCV-Genotyps von 1a zu 1b, ein Befund, der im Grunde beweisend für
eine Neuinfektion ist. Zwei weitere Patienten wiesen im Laufe der
Nachbeobachtung eine positive, aber nicht quantifizierbare HCV-RNA PCR auf,
deren Bestätigungstest noch ausstehend ist. In der Gruppe der Nicht-SVR
Patienten wurden bei 314 Patienten zum Ende der Boceprevir-Tripletherapie RAV
detektiert. Im Verlauf von im Median 24 Monaten nach Therapieende ließ sich
mittels Populations-basierter Sequenzanalyse bei 67% der Patienten (209/314
ausschließlich wieder HCV vom Wildtyp nachweisen (Abb. 5).
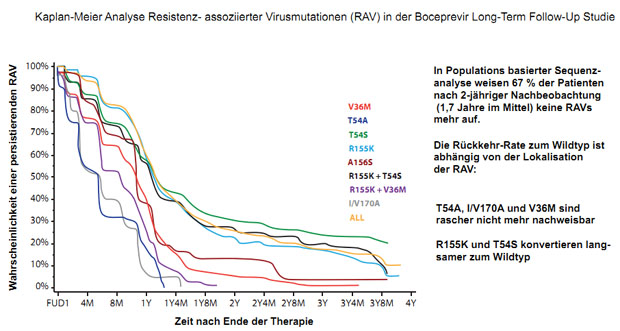 Abbildung 5: Persistenz von
Resistenz-assoziierten HVC-Mutationen nach Boceprevir-Tripletherapie20.
Abbildung 5: Persistenz von
Resistenz-assoziierten HVC-Mutationen nach Boceprevir-Tripletherapie20.
Re-Therapie
Ist eine Re-Therapie nach Telaprevir Versagen möglich? Die bisherigen Studien sprechen dafür, dass die mit einer Proteaseinhibitor assoziierten Resistenzvarianten nach Absetzen der Therapie rasch abnehmen und in den meisten Fällen, zumindest bei der Untersuchung mittels Populations-basierten Sequenzierungsverfahren, sogar vollständig verschwinden. Dass diese Rückentwicklung der Viruspolulation zum Wildtyp aber tatsächlich den erneuten Einsatz von Telaprevir zu einem späteren Zeitpunkt ermöglicht, konnten Sarrazin et al. an einer kleinen Kohorte von 9 Patienten zeigen, die im Schnitt 5,7 Jahre nach Detektion der RAV erneut mit einer Telaprevir-basierten Tripletherapie therapiert wurden. Acht von neun Patienten hatten acht Wochen nach Therapiebeginn HCV-RNA Spiegel unter der Grenze der Quantifizierbarkeit (engl.: limit of quantification, LOQ). Bei sechs dieser Patienten war die HCV-RNA zur Woche 8 negativ. Lediglich ein Patient erlebte einen virologischen Durchbruch, wobei die ursprüngliche Resistenzmutation wieder festgestellt werden konnte21. Leider sind zu dieser Studie keine weiteren Verlaufsdaten publiziert worden.
Die genannte Studie soll abschließend eine wesentliche Frage einleiten, die von Patienten vor Beginn einer Tripletherapie häufig gestellt wird. „Verbaue ich mir möglicherweise Chancen auf zukünftige Therapieoptionen, wenn die Proteaseinhibitor Tripletherapie nicht funktioniert?“ Besonders vor der immer deutlicher sich abzeichnenden Option einer interferonfreien „all oral“ Kombinationstherapie in nicht allzu weiter Zukunft, ist diese Frage relevant. Was ist, wenn im Rahmen eines oralen Therapieregimes ein Proteaseinhibitor eine wesentliche Säule der Wirksamkeit darstellt und diese möglicherwiese aufgrund einer präexistenten Proteaseinhibitorresistenz weg bricht? Mit Daten einer 8-Wochen-Interims-Analyse von 9 behandelten Patienten lassen sich sicher noch keine ausreichend validen Antworten auf diese Frage ableiten. Dennoch, das Prinzip einer wiedererlangten Wirksamkeit aufgrund der Abnahme Resistenz-assoziierter Virusvarianten im Lauf der Zeit ist hiermit erstmals beschrieben worden. Weitere Daten zur Re-Therapie nach Versagen der Proteaseinhibitor Therapie werden mit den großen nicht-interventionellen Studien generiert werden.
- Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011 Jun 23;364(25):2405-16.
- Poordad F, McCone J Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP; SPRINT-2 Investigators. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011 Mar 31;364(13):1195-206.
- Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Müllhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M; REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011 Jun 23;364(25):2417-28.
- Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, Burroughs M, Brass CA, Albrecht JK, Esteban R; HCV RESPOND-2 Investigators. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011 Mar 31;364(13):1207-17.
- C. Hézode, C. Dorival, F. Zoulim, T. Poynard, P. Mathurin, S. Pol, D. Larrey, P. Cacoub, V. de Ledinghen, M. Bourlière, P.H. Bernard, G. Riachi, Y. Barthe, H. Fontaine, F. Carrat, J.-P. Bronowicki, for the CUPIC study group (ANRS CO20). Safety of telaprevir in combination with peginterferon alpha/ribavirin in cirrhotic non reponsers. First results of the French early access program (ANRS CO20-CUPIC). EASL. Apr. 18–22, 2012. oral # 8.
- Colombo M., I. Fernández, D. Abdurakhmanov, P.A. Ferreira, S. Strasser, P. Urbanek, C. Moreno, A. Streinu-Cercel, A. Verheyen, W. Iraqi, R. DeMasi, A. Hill, J.M. Läuffer, I. Lonjon-Domanec, H. Wedemeyer. Treatment of Hepatitis C Genotype 1 Patients with Severe Fibrosis or Compensated Cirrhosis:The International Telaprevir Early Access Program. AASLD 2012, oral. # LB-15
- Berg T., Buggisch P., Hueppe D., Mauss S., Wedemeyer H., Decker-Burgard S. Telaprevir-based triple therapie in patients with chronic hepatitis C in Germany – a 12 weeks interim analysis of real life data. 11th International Congress on Drug Therapy in HIV Infection, 11-15 November 2012, Glasgow, UK
- Forestier N., G. Moog, T. Lutz, M.S. Leuschner, S. Christensen, E. Schott, B. Kallinowski, P. Buggisch, D. Hueppe, U. Alshuth, K. Milicic-Ouakili, S. Mauss. First Real-life data of triple therapy with telaprevir (TVR) and boceprevir (BOC) in combination with peginterferon-alfa-2a (PEG) plus ribavirin (RBV) in patients infected with chronic hepatitis C (CHC), genotype 1 in a non-interventional study (PAN) in Germany. AASLD 2012, abstract #1813
- Mauss S., R. Heyne, T. Lutz, M. Roessle, M. Jung, S. Wollschlaeger, R. Günther, U. Alshuth, D. Hueppe. Safety and week 4 / 12 HCV RNA results of triple combination with telaprevir (TVR)/ peginterferon alfa-2a (P)/ribavirin (R), in F3/F4 patients in real-life setting AASLD 2012, abstract # 1811
- Verna E.C., T. Lukose, S.K. Olsen, A.N. Fox, L.M. Dove, R.S. Brown, N. Terry, K. Mentore, J. Laurin, R. Satoskar, K. Shetty. High Early Response Rates with Protease Inhibitor Triple Therapy in a Multicenter Cohort of HCV-Infected Patients Awaiting Liver Transplantation. AASLD 2012, oral #52
- Hezode C., Y. Barthe, F. Carrat, F. Zoulim, D.G. Larrey, S. Pol, P. Cacoub, V. Canva, T. Poynard, D. Samuel, M. Bourliere, L. Alric, J. Raabe, J.H. Zarski, G. Riachi, P. Bernard, V. de Ledinghen, V. Loustaud-Ratti, S. Metivier, X. Causse, P. Marcellin, J.P. Bronowicki, Safety and efficacy of telaprevir or boceprevir in 455 cirrhotic non responders. Week 16 analysis of the French early access program (ANSR Co20-CUPIC) in real-life setting. AASLD 2012 abstract #51
- Bronowicki J.P., Davis M., Flamm S., Gordon S., Lawitz E., Yohida E., Galati J., Luketic V., McCone J., Jacobson I., Marcellin P., Muir A., Poordad F., Pedicone LD., Deng W., Treitel M., Wahl J. Sustained virologic response (SVR) in prior peginterferon/ribavirin (PR) treatment failures after retreatment with boceprevir (BOC) +PR: the PROVIDE study interim results EASL 2012, oral # 11
- Buti M., Agarwal K., Horsman Y., Sievert W.,Janczewska E., Zeuzem S., Nyberg L., Brown R.S. Hezode C., Rizzetto M., Parana R., De Meyer S., Lou Donghan., Witek J. OPTIMIZE Trial: Noninferiority of twice-daily telaprevir versus adminstration every 8 hours in treatment-naive genotype 1 HCV-infected patients. AASLD 2012 abstract #LB-8.
- Zeuzem S., DeMasi R., Baldini A., Coate B., Luo D., Mrus J., Witek J. Factors Predictive of Anemia Development in Treatment-experienced Patients Receiving Telaprevir (T; TVR) Plus Peginterferon/ribavirin (PR) in the REALIZE Trial. AASLD 2012, abstract #771
- Poordad F.F., Lawitz E.J., Reddy K.R., Afdhal N.H., Hezode C., Zeuzem S., Lee S.S., Lalleja J.L., Brown Jr R.S., Craxi A., Wedemeyer H., Deng W., Koury K., Boparai N., Pedicone L.D., Burroughs M., Wahl J., Brass C.A., Albrecht J.K., Sulkowski M.S. A randomized trial comparing rrbavirin dose reduction versus erythropoietin for anemia management in previously untreated patients with chronic hepatitis C receiving boceprevir plus peginterferon/ribavirin. EASL 2012 abstract #LB-1419
- Poordad F.F., Lawitz E., Reddy K.R., Afdhal N., Hezode C, Zeuzem S., Lee S.S., Calleja J.L., Brown R.S., Craxi A., Wedemeyer H, Bacon B.R., Flamm S.L., Deng W., Koury K.J., Pedicone L., Dutko F., Burroughs M., Alves K., Wahl J., Brass C., Albrecht J.K., Sulkowski M.S. Timing and magnitude of ribavirin dose reduction do not impact sustained virological response rates with boceprevir, peginterferon alpha 2b and ribavirin in the anemia management study in chronic HCV genotyp 1 patients. AASLD 2012 abstract # 154
- Sulkowski M.S., Roberts, S., Afdhal N., Andreone P., Diago M., Pol S., Poordad F.F., Zeuzem S., Bengtsson L., Luo D., Witek J. Ribavirin dose modification in treatment-naïve and previously treated patients who received telaprevir combination treatment: No impact on sustained virological response in phase 3 studies. EASL 2012, abstract #1162
- Sarrazin C., Zeuzem S. Resistance to Direct Antiviral Agents in Patients With Hepatitis C Virus Infection. Gastroenterol 2010;138:447–462
- Zeuzem S, Sulkowski M.S., Zoulim F., and others. Long-term Follow-up of Patients with Chronic Hepatitis C Treated with Telaprevir in Combination with Peginterferon Alfa-2a and Ribavirin: Interim Analysis of the EXTEND Study. AASLD 2010. abstract #227.
- Barnard R.J., Howe A.Y, Hazuda D., Long J., Arja R., Treitel M. Durability of response and analysis of RAVs during long term follow up with boceprevir/peginterferon/ribavirin. An interim analysis. AASLD 2012 abstract #1079
- Sarrazin C., Reesink H.W., Zeuzem S., Weegink C.J., Luo D., Witek J., Kieffer T.L., Bartels D.J., Dierynck I., De Meyer S., Picchio G. Retreatment with Telaprevir/Peg-IFN/RBV after a Short Exposure to Telaprevir in Phase I Studies: Interim Results from a Phase IIIb Rollover Trial (C219). AASLD 2011 oral #35